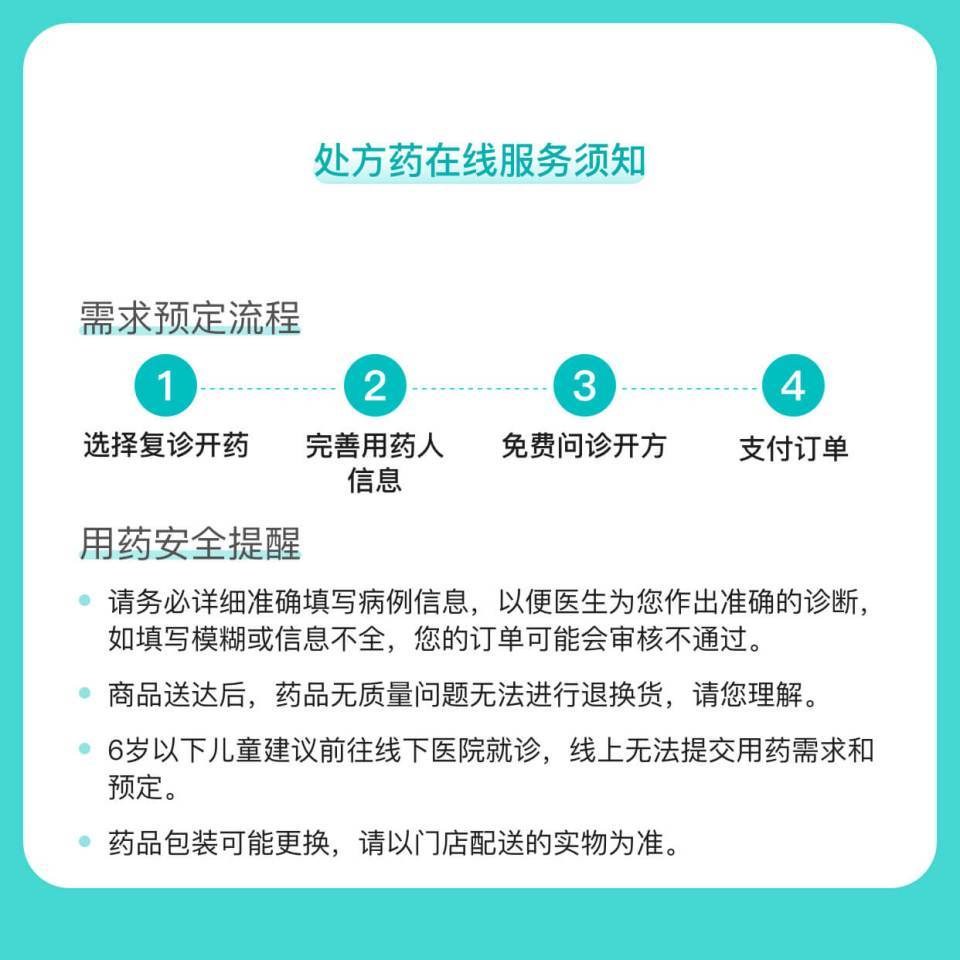
图文介绍







消费者告知
百度健康商城为网络交易服务平台,提供平台技术服务。商家提供商品信息展示、配送、质量安全保障等。
依据《药品经营质量管理规范》,为保障患者用药安全,除药品质量原因外,药品一经售出,不退不换。
依据《药品经营质量管理规范》,为保障患者用药安全,除药品质量原因外,药品一经售出,不退不换。
价格说明
划线价格:指商品的专柜价、吊牌价、正品零售价、厂商指导价或该商品的曾经展示过的销售价等,并非原价,仅供参考。
未划线价格:指商品的实时标价,不因表述的差异改变性质。具体成交价格根据商品参加活动,或使用优惠券等发生变化,最终以订单结算页价格为准
未划线价格:指商品的实时标价,不因表述的差异改变性质。具体成交价格根据商品参加活动,或使用优惠券等发生变化,最终以订单结算页价格为准
如何查询订单?
您可以在百度APP-我的-订单,找到您的商品订单。
如何申请退款?
您可以在百度APP-我的-订单,找到您需要退款的商品订单,在订单详情页选择“退款”,按照指引流程进行操作即可。退款成功后,您的资金将原路返回您的支付账号,因不同银行处理时间不同,预计1~5个工作日内到账。
如何查找小程序?
打开百度 App,搜索「百度健康商城」即可直达小程序
商品评价(1234)
查看全部

c***6

30mg*7片/盒
#正品#实惠#质量好效果很不错价格比药店便宜多了,同一个品牌在药店里买要贵多了,性价比高,吃了以后办个小时才能发挥药效,效果你买了就知道😁😁。客服回复也很及时,告诉你什么一些药物使用的禁忌。



1***5
30mg*7片/盒
带劲,之前主要问题是硬度不够,夫妻之间难以满足,有时为了延长时间,忍着不射,但是时间一长很快软了,更难满足老婆。所以疲软就需要达泊西丁,服用一粒,不够的话可以适当调整,大概半小时后,稍微刺激一下就能感觉下面很硬,跟老婆做了四十分钟,都很满足!有那么一点副作用吧,多喝水就可以了,效果是真的很好,会推荐的




h***6
30mg*7片/盒
#正品#实惠#质量好#效果不错#图文相符#包装严实#物流快#回复很快达泊西汀吃了之后最直接的感觉就是龟头不敏感了,但与延时喷剂麻木感不一样,还是有感觉的,对于延时确实有效果,不需要刻意转移注意力,女友不算那种欲望很强的人,换了两个姿势说累,我觉得可以尝试一下吃达泊西汀延时,




1***2
30mg*7片/盒
#正品#实惠#质量好#效果不错#图文相符#包装严实#物流快#回复很快质量值得信赖,与描述的商品效果一样,此商家的商品绝对正品,价格合理性价比很高,值得再次购买,我已是第二次回购,希望以后的日子里,能有更好的商品,回馈广大客户。
1***9
30mg*7片/盒
#正品#实惠#质量好#效果不错#图文相符#包装严实#物流快#回复很快正品实惠质量很好很好,效果不错不错,图文相符,包装完好无损!

不***Z
30mg*7片/盒
#物流快用着感觉很不错,效果好,物流快,保密性好,客服回复快,经济实惠,下次还会买
看了又看






店铺推荐




