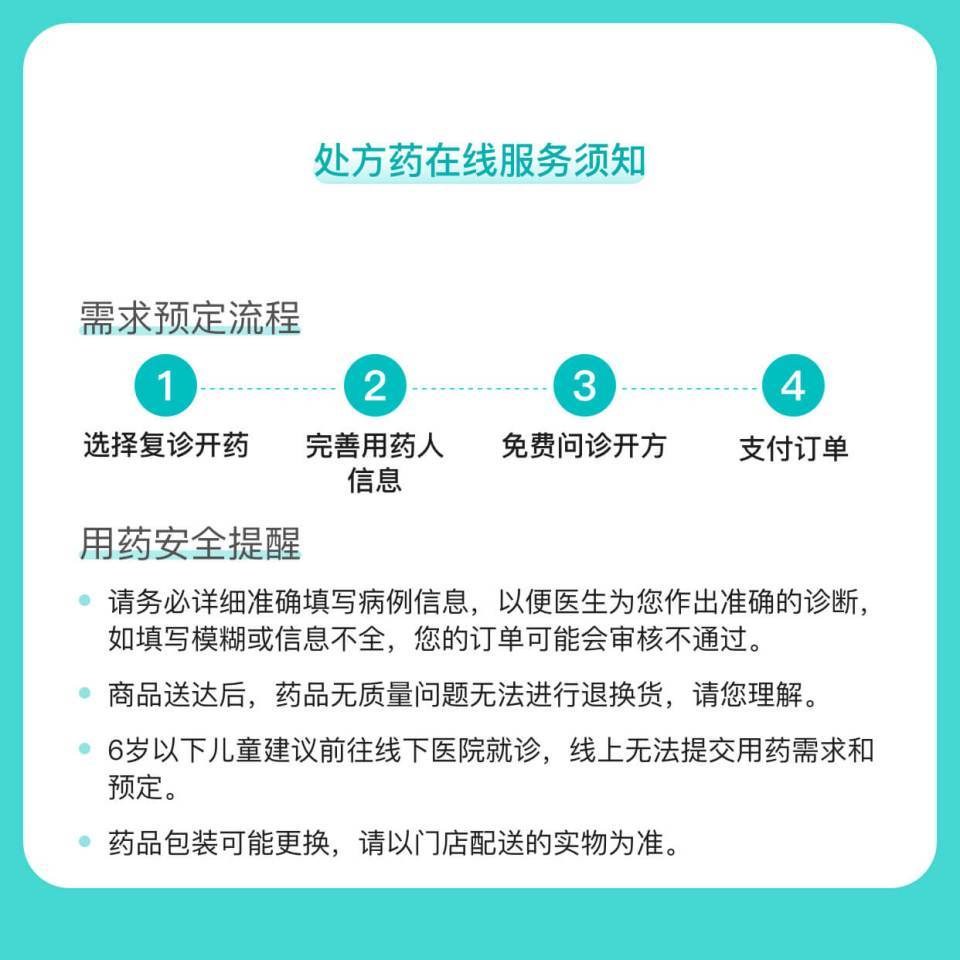
图文介绍


消费者告知
百度健康商城为网络交易服务平台,提供平台技术服务。商家提供商品信息展示、配送、质量安全保障等。
依据《药品经营质量管理规范》,为保障患者用药安全,除药品质量原因外,药品一经售出,不退不换。
依据《药品经营质量管理规范》,为保障患者用药安全,除药品质量原因外,药品一经售出,不退不换。
价格说明
划线价格:指商品的专柜价、吊牌价、正品零售价、厂商指导价或该商品的曾经展示过的销售价等,并非原价,仅供参考。
未划线价格:指商品的实时标价,不因表述的差异改变性质。具体成交价格根据商品参加活动,或使用优惠券等发生变化,最终以订单结算页价格为准
未划线价格:指商品的实时标价,不因表述的差异改变性质。具体成交价格根据商品参加活动,或使用优惠券等发生变化,最终以订单结算页价格为准
如何查询订单?
您可以在百度APP-我的-订单,找到您的商品订单。
如何申请退款?
您可以在百度APP-我的-订单,找到您需要退款的商品订单,在订单详情页选择“退款”,按照指引流程进行操作即可。退款成功后,您的资金将原路返回您的支付账号,因不同银行处理时间不同,预计1~5个工作日内到账。
如何查找小程序?
打开百度 App,搜索「百度健康商城」即可直达小程序
看了又看






店铺推荐





